
1. がん治療薬
1-1. ユビキチン-プロテアソームシステムに対する阻害作用
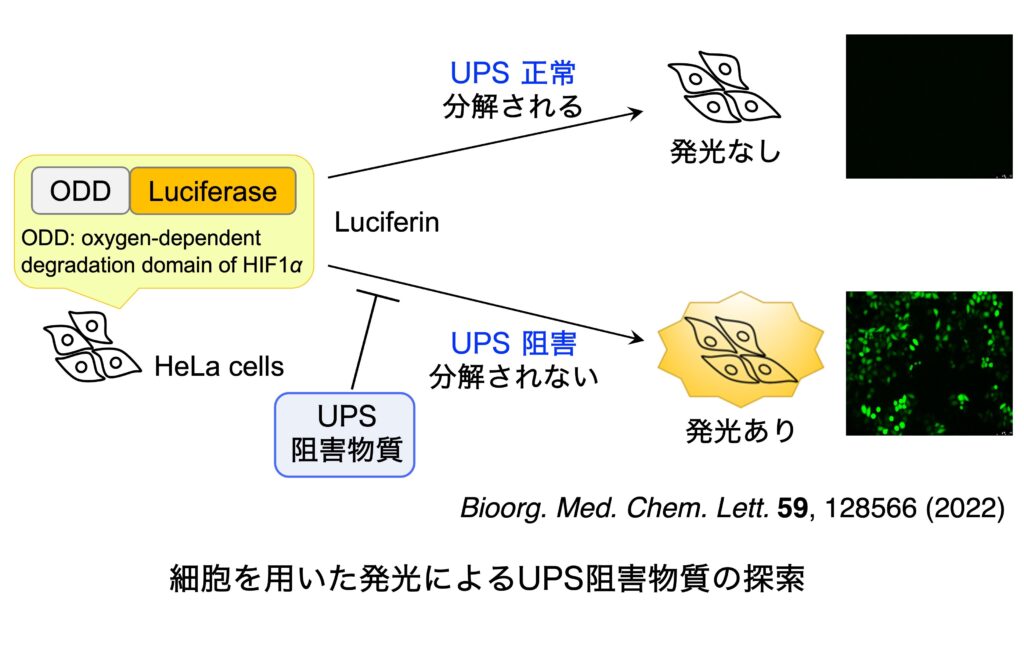
ユビキチン-プロテアソームシステムは、ユビキチン化を触媒するユビキチンシステムと、タンパク質分解装置である 26Sプロテアソームから構成されています。そして、生体内において分解される運命にあるタンパク質は、ユビキチンシステムによりポリユビキチン化された後(ステップA〜D)、26Sプロテアソームへとリクルート(ステップE)され、脱ユビキチン化(ステップF)の後に ATP 依存的に分解されます(ステップG)。ユビキチン-プロテアソームシステムは、細胞周期の進行、シグナル伝達、遺伝子発現制御、タンパク質の品質管理など、多様な生命現象に関与しています。したがって、この分解系の破綻は疾病につながり、また一方では、この分解系が創薬の標的となります。2003年に、プロテアソーム阻害物質である Bortezomib が多発性骨髄腫の治療薬としてアメリカで認可され、現在では世界中で使用されています。そして引き続き2012年には、Bortezomib に 耐性な多発性骨髄腫に有効なプロテアソーム阻害剤として Carfilzomib が、そして2015年11月には経口投与可能なプロテアソーム阻害剤である Ixazomib が認可されました。また、海洋微生物から発見された salinosporamide A (Marizomib) については Phase III の臨床試験が行われています。現在、プロテアソームに加えて、その上流で翻訳後修飾ユビキチン化を司るユビキチンシステム(E1、E2、E3酵素からなる)やその逆反応を触媒する脱ユビキン化酵素も創薬の標的として注目を集め、ユビキチン-プロテアソームシステムの全体を標的とする創薬研究が活発に行われています。しかしその研究は、化合物ライブラリーからの探索や、リード化合物の修飾による方法で行われており、天然資源からの探索はあまり行われていないません。そこで私たちは、Bortezomib が認可される以前から、構造的に多様な低分子化合物の宝庫である天然資源を対象として、ユビキチン-プロテアソームシステムに対する阻害物質の探索を行ってきました。最近は、ルシフェラーゼが融合した ODD を発現する細胞を用いて発光により検出する方法を用いて、UPS 阻害物質を探索しています。また、UPS 阻害物質の探索において、がん抑制遺伝子産物である p53 タンパク質の分解を阻害し、その結果、抗がん作用を示すことが期待できるような化合物の探索を重点的に行っていましたが、最近は、がん組織において p53 の半数が変異していることに注目し、変異型 p53 を標的とする化合物の探索を行っています。
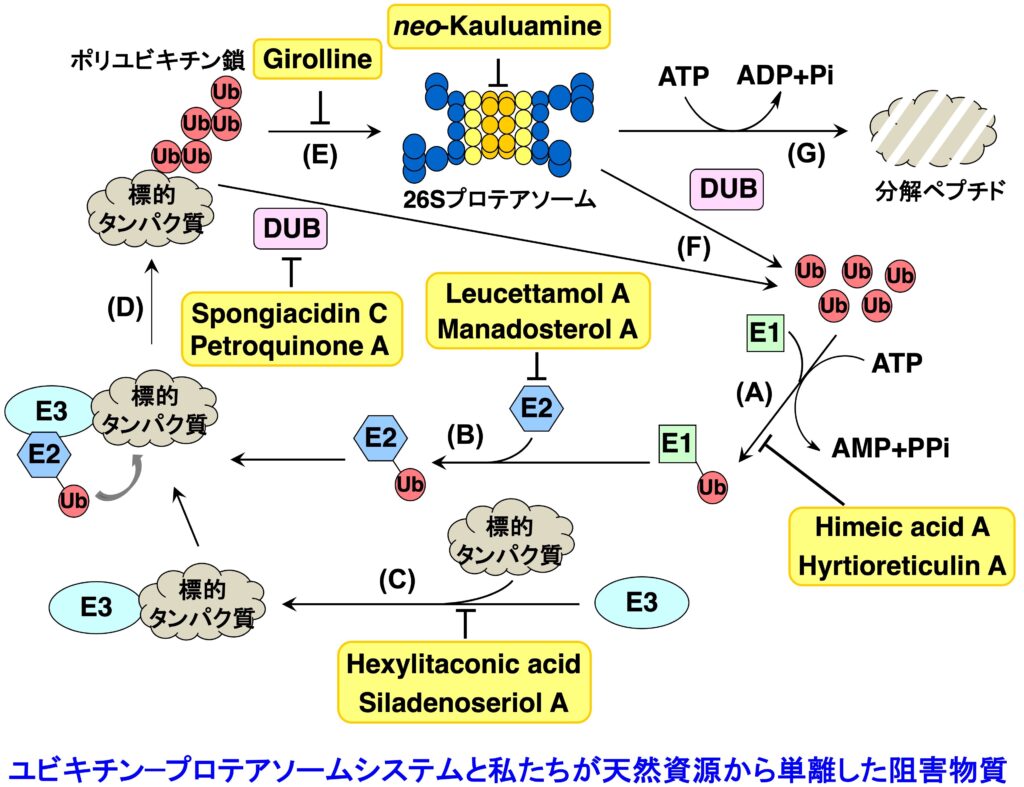
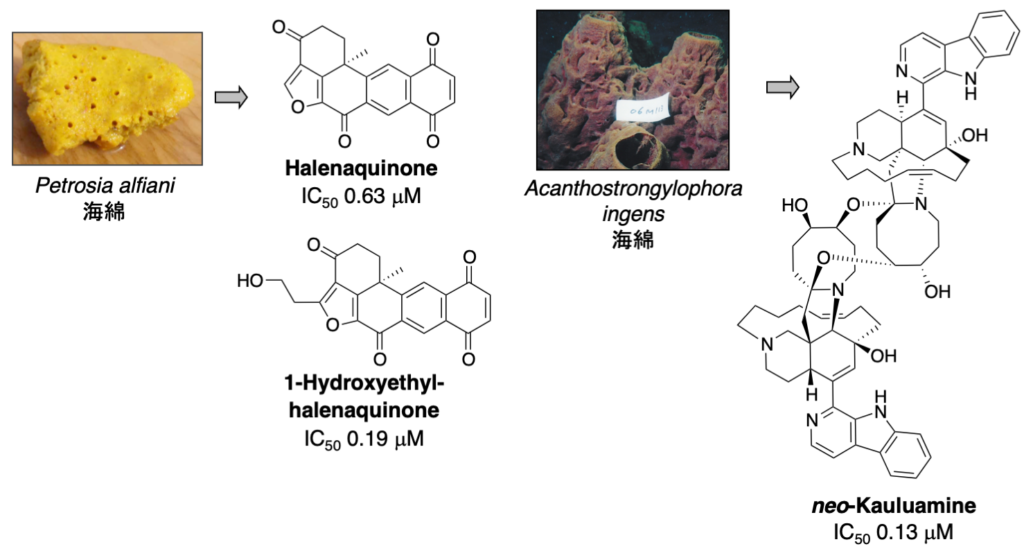
1-1-1. プロテアソーム阻害物質 (ステップG)
プロテアソームは細胞内に普遍的に存在しますが、特にがん細胞で活性が高いことが知られています。プロテアソーム阻害剤の開発は活発に行われていますが、その研究は合成化合物からのスクリーニングあるいはリード化合物の修飾によるものがほとんどです。私たちは、天然資源から新規骨格を有するプロテアソーム阻害物質を発見するため、天然資源抽出物をスクリーニングし新規プロテアソーム阻害物質を探索しています。特に、免疫型プロテアソームも含め、サブユニット特異的阻害物質の発見を目指しています。
1. Mellanin A: Bioorg. Med. Chem. Lett. 59, 128566 (2022)
2. Halicyclamine B: Bioorg. Med. Chem. Lett. 29 (1), 8-10 (2019).
3. Strongylophorines: Bioorg. Med. Chem. Lett. 25 (13), 2650-2653 (2015).
4. neo-Kauluamine: J. Nat. Prod. 77 (6), 1536-1540 (2014).
5. 1-Hydroxyethylhalenaquinone: Heterocycles 89 (11), 2605-2610 (2014).
6. Salsolinol: Chem. Pharm. Bull. 59 (2), 287-290 (2011).
7. Aaptamine: Bioorg. Med. Chem. Lett. 20 (11), 3341-3343 (2010).
8. Lignans from Anemarrhenae Rizoma: Biol. Pharm. Bull. 28 (9), 1798-1800 (2005).
9. Secomycalolide A: Mar. Drugs 3 (2), 29-35 (2005).
10. Agosterol C: J. Nat. Prod. 66 (12), 1578-1581(2003)
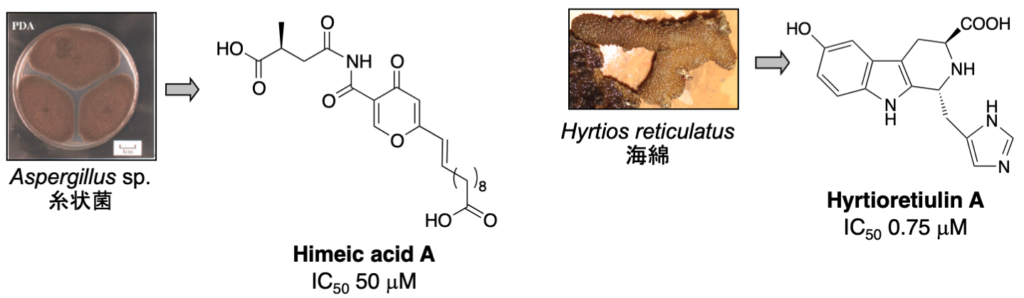
1-1-2. ユビキチン活性化酵素 (E1) 阻害物質 (ステップA)
E1, E2, E3 酵素から構成されるユビキチンシステムのいずれかを阻害した場合にも 26Sプロテアソームによるタンパク質分解は阻害されますので、プロテアソームを阻害した場合と同様に抗がん剤を発見できると期待されます。私たちは、E1 阻害物質を探索するための評価系を確立し新規阻害物質 himeic acid A を発見しました。E1を阻害する化合物としては、世界で2 例目に発見された化合物となります。さらに、より阻害活性の強い化合物として hyrtioreticulin A を海綿から単離しました。
1. Hyrtioreticulin A: Bioorg. Med. Chem. 20 (14), 4437-4442 (2012).
2. Himeic acid A: Bioorg. Med. Chem. Lett. 15 (1), 191-194 (2005).
1-1-3. ユビキチン結合酵素(E2)阻害物質 (ステップB)
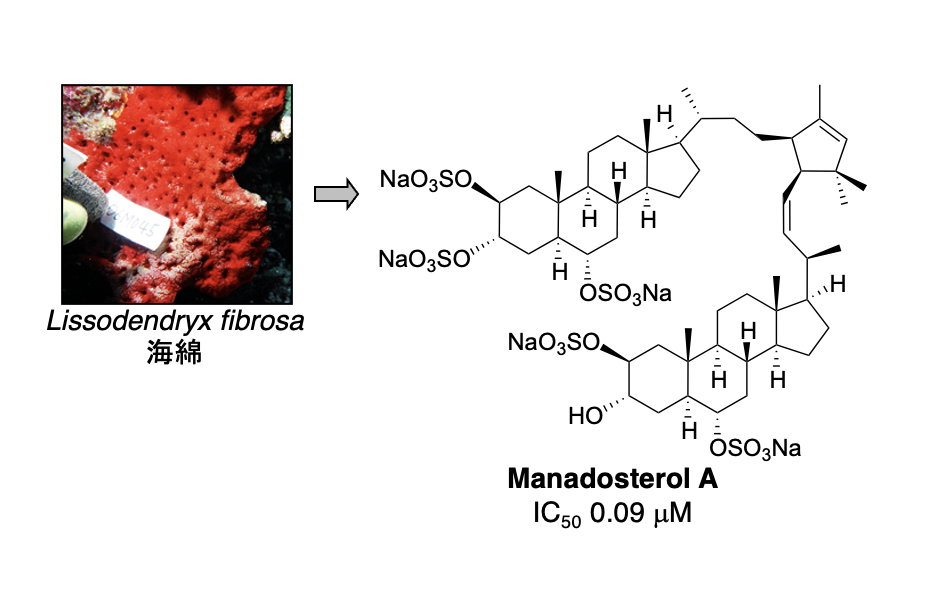
E2 は約40種類存在しますが、その中でも Ubc13 は Uev1A とヘテロ複合体を形成して E2 酵素として働きます。そして、生合成中の p53 に Ubc13-Uev1A 複合体が結合すると、p53 の K63-ユビキチン化が起こり発がんに至ることが報告されています。したがって、Ubc13-Uev1A 複合体形成を阻害する化合物は、がん抑制作用を示すと考えられます。私たちは、Ubc13-Uev1A 複合体の形成を検出する評価系を確立し、世界初の E2 阻害物質として海綿から leucettamol A を単離することに成功しました。その後、より阻害作用の強い化合物として manadosterol A を海綿から単離しました。
1. Manadosterol A: J. Nat. Prod. 75 (8), 1495-1499 (2012).
2. Leucettamol A: Bioorg. Med. Chem. Lett. 18 (24), 6319-6320 (2008).
1-1-4. ユビキチンリガーゼ (E3) 阻害物質 (ステップC)
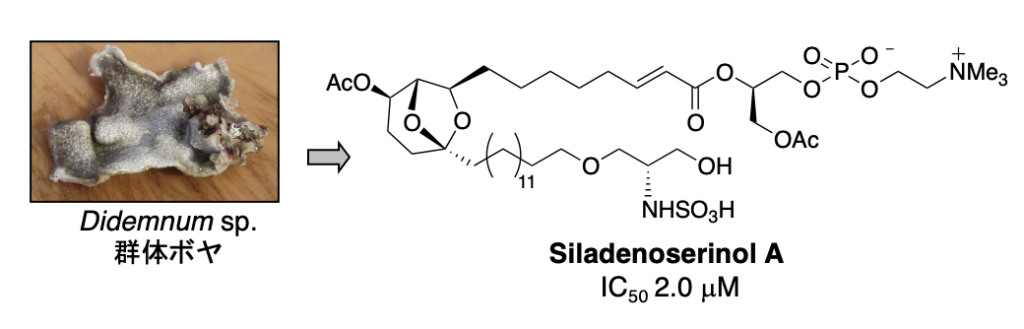
プロテアソームによって選択的にタンパク質が分解される際、生体内に存在する多くのタンパク質の中から分解すべきタンパク質を認識するのは E3 です。E3 は約600種類存在すると言われていますが、私たちは Mdm2 に注目しました。Mdm2 は、1997年にがん抑制遺伝子産物 p53 の E3 であることが明らかにされました。このことから、Mdm2 拮抗剤は p53 に対する負の調節因子である Mdm2 の作用を阻害するだけでなく、p53 の分解を阻害することにより、生体内における p53 レベルを上昇させると考えられます。したがって、Mdm2 拮抗剤は、p53 のがん抑制タンパク質としての機能である「細胞修復」や「アポトーシス」を亢進させる新しいタイプのがん治療薬として期待できます。合成化合物のライブラリーから発見された Nutlin は、in vivo でもがん縮小効果を示しましたが、天然資源からの探索研究はほとんど行われていません。私たちは、p53-Mdm2 複合体形成を阻害する化合物として、海綿由来の真菌から hexylitaconic acid を単離しましたが、その後、群体ボヤから siladenoserinol A と命名した新規阻害物質を発見することに成功しました。
1. Niphateolide A: Tetrahedron 71 (38), 6956-6960 (2015).
2. Siladenoserinol A: Org. Lett. 15 (2), 322-325 (2013).
3. Hexylitaconic acid: Bioorg. Med. Chem. Lett. 16 (1), 69-71 (2006).
1-1-5. USP7阻害物質 (ステップF)
Mdm2 は自らをポリユビキチン化してプロテアソームにより分解されます。この時、脱ユビキチン化酵素 USP7 は Mdm2 に結合して自己ユビキチン化された Mdm2 からユビキチンを除去する働きをしているので、結果として p53 の分解が起きます。したがって、USP7 に対する阻害物質は Mdm2 の分解を誘導することにより p53 を安定化させるので、選択性の高いがん治療薬になると考えられます。私たちはスクリーニングを行い、海綿から spongiacidin C や petroquinone A を単離しました。
1. 2-Geranyl-2′,3,4,4′-tetrahydroxydihydrochalcone: J. Nat. Med. 72 (3), 632-640 (2018).
2. Sulawesins A: J. Nat. Prod. 80 (7), 2045-2050 (2017).
3. Petroquinone A: Tetrahedron 72 (35), 5530-5540 (2016).
4. Spongiacidin C: Bioorg. Med. Chem. Lett. 23 (13), 3884-3886 (2013).

1-2. 変異型 p53 を標的とする創薬研究
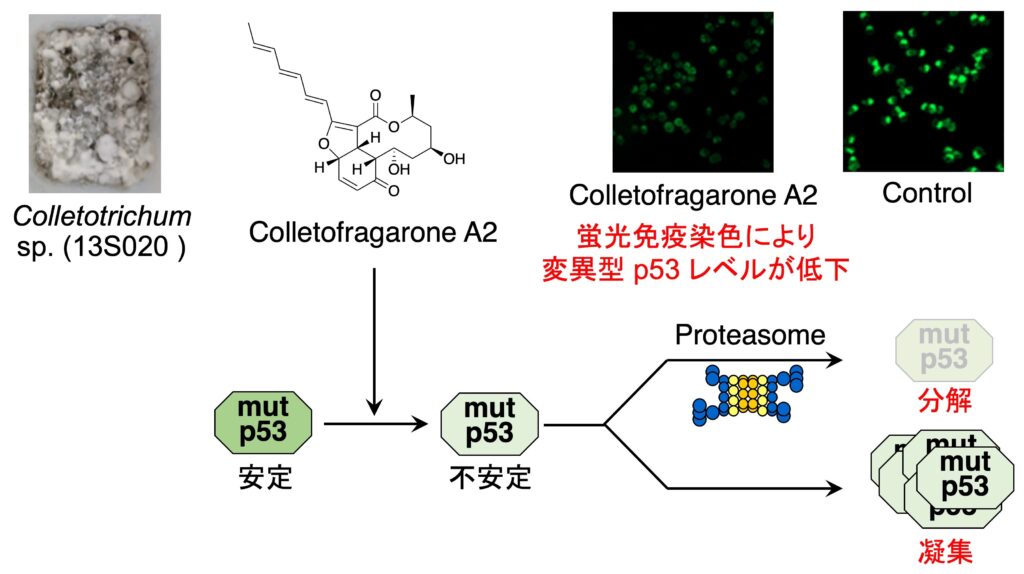
がん組織では、半数の p53 が変異していると言われています。そして、変異した p53 は凝集しがんの進行や浸潤、転移を亢進させることが知られています。そこで、私たちは、変異型 p53 を野生型様へと機能回復させることのできる天然物を探索するため、蛍光免疫染色によるスクリーニングを行いました。そして、真菌から変異型 p53 レベルを減少させる化合物として colletofragarone A2 を発見しました。Colletofragarone A2 は、変異型 p53 を有するがん細胞を移植したマウスに対して抗腫瘍作用を示しましたが、作用について解析したところ、 変異型 p53 のプロテアソームによる分解と凝集を誘導させることが明らかとなりました。
1. Colletofragarone A2: J. Nat. Prod. 84 (12), 3131-3137 (2021); Chem. Res. Toxicol. 35 (9), 1598-1603 (2022) .
1-3. 細胞周期阻害物質
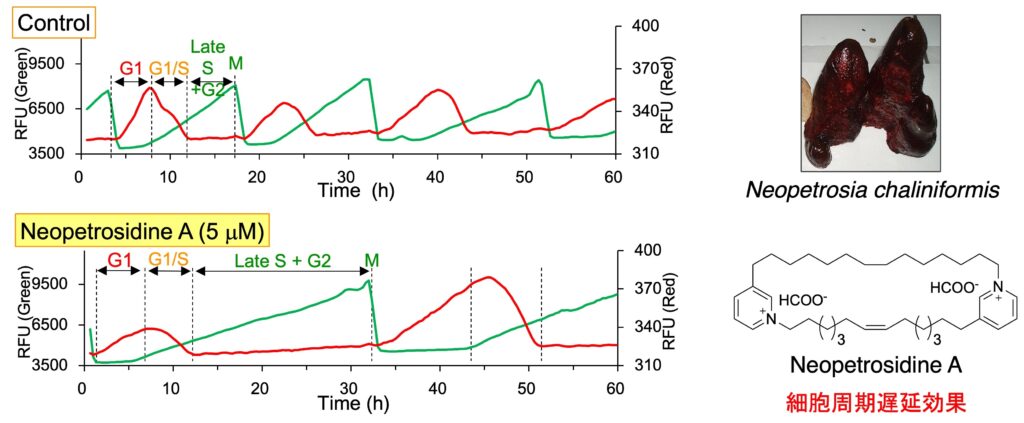
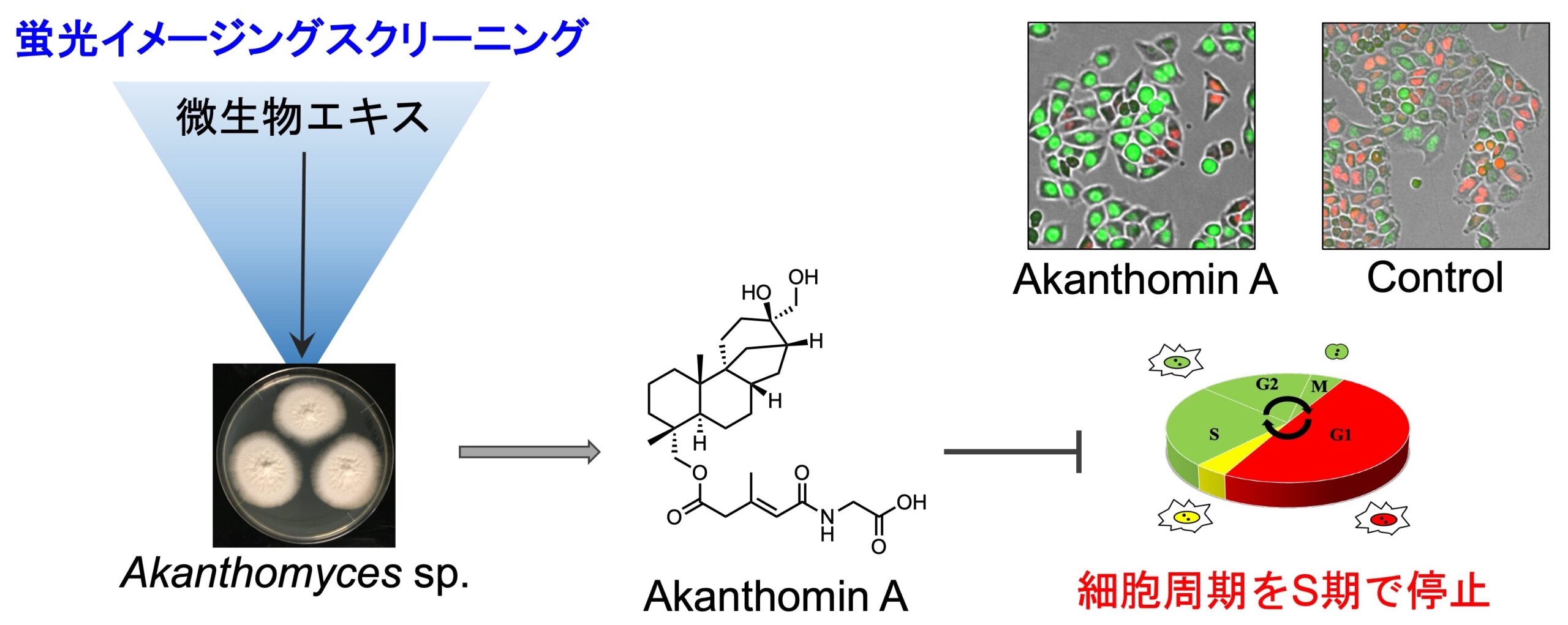
細胞周期依存的に発現・分解するタンパク質に緑色・赤色蛍光タンパク質を融合した細胞周期可視化マーカー Fucci(Miyawaki, A. 2008, 2011)を有する HeLa/Fucci2 細胞を用いると、生細胞の状態で経時的な細胞周期の進行を観察することができます。この細胞を用いてハイスループットな表現系スクリーニングを行い、細胞周期を停止させる化合物あるいは細胞の形態を変化させる化合物を探索しています。海綿 Dactyspongia metachromia から、S/G2/M 期で細胞周期を停止させる新規化合物 neoisosmenospongine を発見しました。また、海綿 Neopetrosia chaliniformis から単離した新規化合物 neopetrosidine A は、当初、G1期で細胞周期を停止させると思われましたが、タイムラプス解析により細胞周期を遅延させることがわかりました。
1. Akanthomin A: Phytochemistry 216, 113885 (2023).
2. Neopetrosidine A: Bioorg. Med. Chem. 50, 116461 (2021).
3. Neoisosmenospongine: Bioorg. Med. Chem. 31, 115968 (2021).
4. Girolline: Biol. Pharm. Bull. 27 (5), 691-701 (2004).

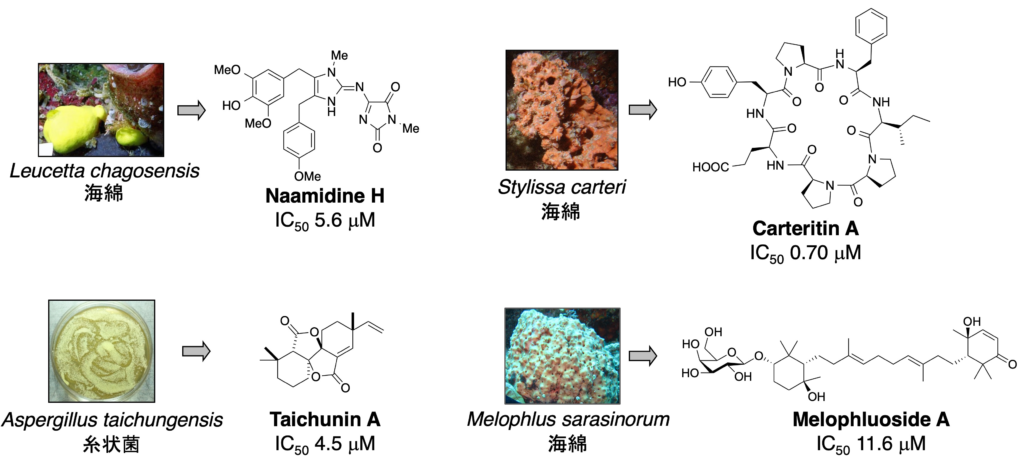
1-4. がん細胞に対する増殖抑制作用
1. Melophluoside A: Tetrahendron Lett. 61 (20), 151852 (2020).
2. Taichunin A: J. Nat. Prod. 82 (5), 1377-1381 (2019).
3. Carteritin A: Tetrahedron Lett. 57 (11), 1285-1288 (2016).
4. Naamidines H & I: J. Nat. Prod. 70 (10), 1658-21660 (2007).
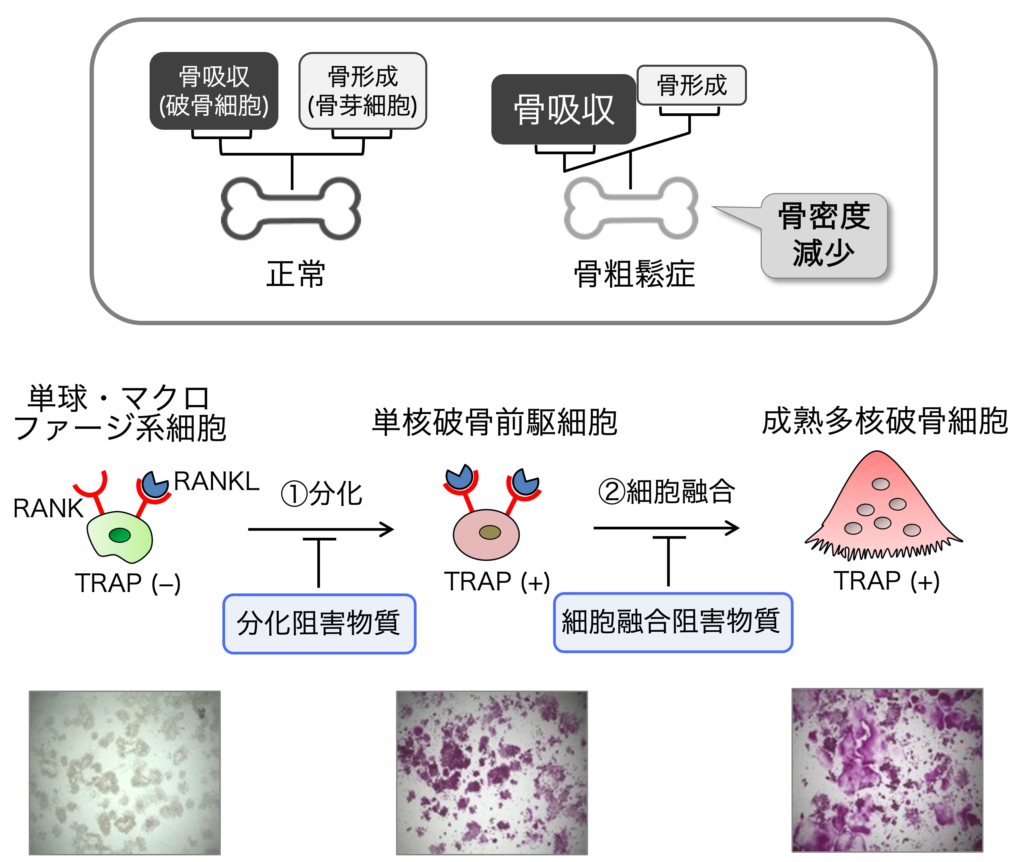
2. 骨粗鬆症治療・予防薬
2-1. 多核破骨細胞形成に対する阻害作用
骨は新陳代謝をしており、破骨細胞による骨吸収と骨芽細胞による骨形成のバランスが維持されることにより、健康な骨が維持されます。そして骨粗鬆症は、骨吸収の亢進により骨量が減少し骨折の危険性が増大する疾患です。私たちは、多核破骨細胞の形成を阻害する成分を天然資源から探索しています。
1. Aaptocarbamates A−G: Phytochemistry 216, 113872 (2023).
2. Arteperoxides A–C: Phytochemistry 206, 113548 (2023).
3. Amakusamine: J. Nat. Prod. 84 (10), 2738-2743 (2021).
4. Taichunins E–T: J. Nat. Prod. 84 (9), 2475-2485 (2021).
5. Aaptic acid: Heterocycles 97 (2), 1219-1225 (2018).
6. Neviotine D: Fitoterapia 128, 43-49 (2018).
7. Ceylonin A: J. Nat. Prod. 80 (1), 90-95 (2017).
8. (-)-6-epi-Notoamide T: Bioorg. Med. Chem. Lett. 27 (22), 4975-4978 (2017).
9. Ceylonamide A: J. Nat. Prod. 79 (8), 1922-1928 (2016).
10. Halenaqionone: Bioorg. Med. Chem. Lett. 24 (22), 5315-5317 (2014).
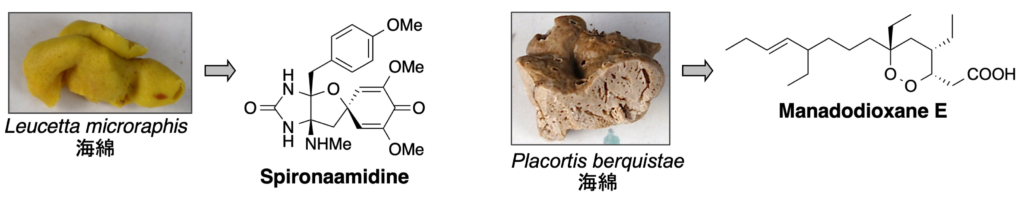
3. 感染症治療薬
3-1. 抗菌作用
1. Manadodioxane E: J. Nat. Med. 69 (4), 595-600 (2015).
2. Spironaamidine: Tetrahedron Lett. 52 (41), 5342-5344 (2011).
3-2. HIV 潜伏感染細胞に対する活性化物質
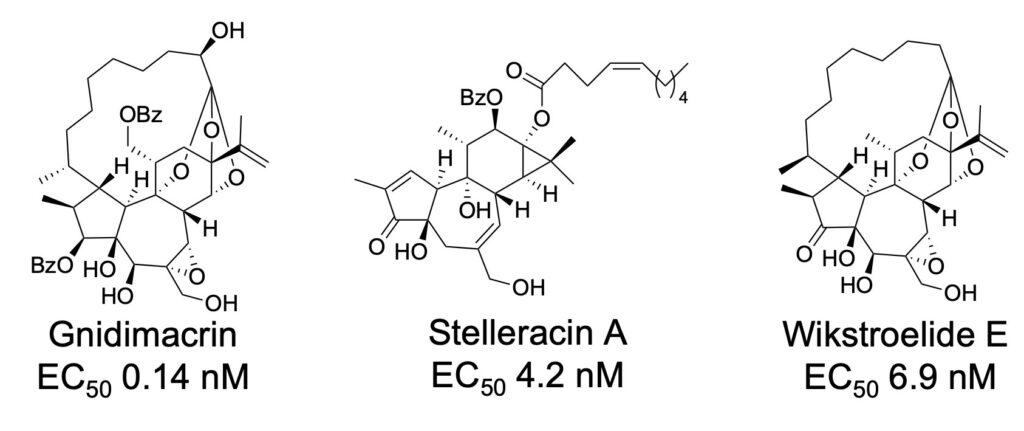
エイズは、抗ウイルス薬の服用により感染しても発症を抑えることが可能になりましたが、潜伏感染している HIV を除去できないため、一生抗ウイルス薬を服用する必要があります。そこで、潜伏感染細胞に対する活性化物質(LRA)を探索し、イモガンピ(ジンチョウゲ科植物)からgnidimacrin(EC50 0.14 nM)などのジテルペンを単離しました。また、抗 HIV 薬(EFdA)と既存の LRA(PEP005)の併用では休薬後にリバウンドしましたが、stelleracin A や wikstroelide E との併用ではリバウンドせず、さらに 単独でも HIV が検出限界まで減少しリバウンドしないことを見出しました。
J. Med. Chem. 65 (4), 3460-3472 (2022).
4. 創薬研究に関する総説
1. Y. Hitora, S. Tsukamoto. The search for inhibitors of the ubiquitin-proteasome system from natural sources by cell-based screening in reporter-expressing cells. J. Synth. Org. Chem., Jpn. 81 (11) 1073-1080 (2023).
2. El-Desoky, A. H. H., Tsukamoto, S. Marine natural products that inhibit osteoclastogenesis and promote osteoblast differentiation. J. Nat. Med. 76, 575-583 (2022).
3. 人羅勇気、塚本 佐知子. p53を標的とするがん治療薬. 実験医学増刊「ゲノム医療時代のがん分子標的薬と診断薬研究(編集:西尾和人)」、羊土社、38 (15), 184-190 (2020).
4. 加藤光、塚本 佐知子. 計算化学を活用した天然有機化合物の構造解析.化学工業、70 (10),41-45 (2019).
5. Tsukamoto, S. Search for inhibitors of the ubiquitin-proteasome system from natural sources for cancer therapy. Chem. Pharm. Bull. 64 (2), 112-118 (2016).
6. 塚本佐知子、横沢英良. ユビキチン修飾系を標的とする創薬シーズの探索. 実験医学増刊「シグナル伝達 研究最前線2012」、編集:井上純一郎、武川睦寛、徳永文稔、今井浩三、羊土社、30 (5), 171-176 (2012).
7. 塚本佐知子、横沢英良. ユビキチン依存的タンパク質分解系を標的とする創薬. 化学と生物、 49 (11), 745-754 (2011).
8. 塚本佐知子、横沢英良.タンパク質分解の不思議:ユビキチン‐プロテアソームシステムの仕組みと創薬.愛知学院大学薬学会誌 3, 1-18 (2010).
9. Tsukamoto, S., Yokosawa, H. Inhibition of the Ubiquitin-proteasome System by Natural Products for Cancer Therapy. Planta Med. 76 (11), 1064-1074 (2010).
10. Tsukamoto, S., Yokosawa, H. Targeting the proteasome-mediated proteolytic pathway, Expert Opin. Ther. Targets 13 (5), 605-621 (2009).
11. 塚本佐知子. ユビキチンリガーゼを分子標的とする新規抗がん剤の海洋生物からの探索. 薬学研究の進歩研究成果報告集、24, 45-51 (2008).
12. 塚本佐知子、横沢英良. 展開するプロテアソーム阻害剤研究. 実験医学増刊「細胞内の輪廻転生 タンパク質の分解機構」、編集:田中啓二、羊土社、26, 122-127 (2008).
13. Tsukamoto, S. The Search for Inhibitors of the Ubiquitin-proteasome System from Natural Resources for Drug Development. J. Nat. Med. 60 (4), 273-278 (2006).
14. Tsukamoto, S., Yokosawa, H. Natural Products Inhibiting the Ubiquitin-proteasome Proteolytic Pathway, a Target for Drug Development. Curr. Med. Chem. 13 (7), 745-754 (2006).
15. 塚本佐知子、横沢英良. ユビキチン-プロテアソームシステムをターゲットとする天然物化学・創薬化学の新しい展開. 有機合成化学協会誌、62, 968-976 (2004).
